(10-07-2023, 11:42 PM)AimSmall Wrote: Just anything you learn would be interesting. It’s a reason many of us are here is to learn, be it analysis results or techniques.
I am currently looking for some new methods that could help in the analysis of populations or individuals. Inspired by this thread in particular:
https://genarchivist.com/showthread.php?tid=110 . I tried to carry out clustering with the help of plink of some samples of Ukrainians that I had in plink format, and compared with the results on G25. At the moment, it is still difficult for me to interpret the results and find patterns. During clustering, samples that were previously considered outliers (EG600048 and EG600093) were filtered out, which is good. Also, samples EG600037 and EG600038 were very close, next to each other, they come from the same village (they are very far from each other by G25 distances). Further, I have not yet found any patterns of division into clusters. Tried the function
IBS similarity matrix https://zzz.bwh.harvard.edu/plink/strat.shtml#matrix , looked at the distances between populations, but those populations that are closest to each other on the distance matrix may not be located on the same branch during clustering.
By the way, do you know the method, how can I directly compare two samples to each other? Maybe this IBS similarity matrix is just for that? I mean some analogue of distances for calculators, only directly comparing the genotype. For example, the difference between alleles between two individuals. I tried
f2 and
fst in admixtools 2, but firstly, these functions are non-linear, secondly, their results vary greatly depending on the number of samples that are compared at the same time, and I'm not sure that I'm doing everything correctly, so they gave some faint results only for long distances, that is, if you compare, for example, modern samples with some ancient hunters.
Are there any analogues of f2 statistics in the same PLINK? For example, I have the distance from a sample to other samples on the G25 calculator, and I would like to see how correct these distances are, that is, to directly compare the genotypes of these samples, for example, by allele frequency. How can this be done? Is this IBS similarity matrix suitable for this?
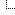


![[Image: PLINK.jpg]](https://www.imgkub.com/images/2023/10/07/PLINK.jpg)